|
||
DMSO Background Literature |
|||
|
Claudio B. Caputa and Andre I. Salama Department of Pharmacology • ICI Pharmaceuticals Group • ICI Americas Inc. • Wilmington, DE 19897 Received 21 February 1989
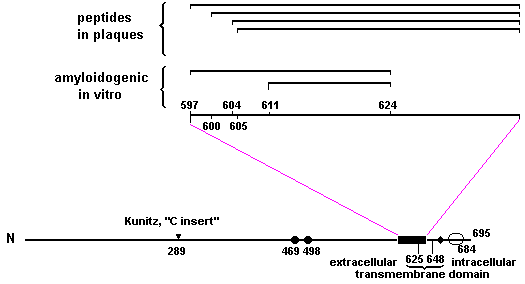
It is plausible that certain posttranslational events may render proteins amyloidogenic in vivo. This concept has attracted recent attention among investigators interested in the amyloid of Alzheimer's disease. While several studies have failed to provide evidence for a transcriptional problem,31,89,118,124 other studies suggest that numerous posttranslational events occur to the proteins that comprise plaques and tangles (see below). Should any of these events be essential for amyloid production, then correcting them may provide a therapeutic means to correct amyloid accumulation. An advantage of this approach is that many such events are mediated through enzymes, and enzymes have provided very effective therapeutic targets for other diseases. 60,88 Some of the more thoroughly studied posttranslational events relevant to Alzheimer proteins are considered here. Proteolysis Substantial evidence suggests that proteolysis may be a mechanism for amyloid formation. The amyloid deposits associated with myeloma are derived from Bence Jones proteins, the immunoglobulin light chain dimers that are synthesized by the malignant plasma cells in this disease. 35 While it has not been established that proteolysis of these proteins in vivo is responsible for the amyloid associated with myeloma, it has been shown that the soluble Bence Jones proteins form amyloid in vitro as a result of proteolysis. 38 Several proteases produce this effect, including trypsin, pepsin, and lysosomal enzymes. 28,69,112 In Alzheimer's disease, beta amyloid protein is deposited in the walls of vessels and in extracellular parenchyma in the brain. 140 This amyloid protein has been solubilized, its sequence determined,39,75 and the gene for it cloned. 42,61,95,125 The precursor contains 695 amino acid residues,61 which is substantially longer than the amyloid products of up to 42 amino acids (Fig. 2), suggesting that proteolysis has occurred. The various amyloid products share the same carboxyl teminus (amino acid 638 of the precursor) while their amino termini include four sites (amino acids 597, 600, 604, and 605). Synthetic peptides comprised of at least amino acids 611 to 624 and up to 597 to 624 of the precursor (see Fig. 2) have been shown to form amyloid fibrils spontaneously in vitro with physical characteristics that closely resemble the fibrils isolated from Alzheimer brains. 17,65 Peptides with less than this minimum fourteen amino acid sequence (amino acids 611 to 624) formed fibrils with different physical characteristics. Although some protease cleavage may also occur after amyloid formation,128 this cleavage would not be an appropriate drug target.
FIG 2. The 695 amino acid beta amyloid protein precusor (61) is shown with three potential sites for N-glycosylation indicated by circles, two of which are likely to be glycosylated [solid circles6l], two potential phosphorylation sites indicated with a diamond,32 and the site where a Kunitz inhibitor-like sequence of amino acids is variably inserted either alone to yield the 751 amino acid version of the precursor or in combination with an adjacent sequence (on the C terminal side of the inhibitor "C insert") to yield a 770 amino acid version. 67,90,126 The beta amyloid protein sequence is indicated with a bar and shown in more detail above the precursor. The four beta amyloid protein sequences that have been identified in neuritic plaques75 are shown, as well as sequences that form amyloid fibrils in vitro,17,64 including the minimum amyloidogenic sequence of amino acids 611 to 624. 17 Although no protease has yet been shown to produce beta amyloid protein, a number of leads exist to help direct an empirical search for such proteases. For example, elastase, a serine protease, is known to be bound to various amyloids, as mentioned above. In addition, plaques and tangles have been shown to contain cholinesterases which have potent serine protease activity. 78 Ubiquitin, which is present in PHF-containing plaques (associated with the PHFs), recently has been shown to possess serine protease activity. 18,20,30 Levels of the aspartyl protease, cathepsin D, were reported to increased in Alzheimer brains compared to normal senile brains,123 as well as in aging in the absence of disease. 77 Two protease inhibitors have been linked to beta amyloid protein, although no functional role for them in amyloid formation has yet been established. The serine protease inhibitor alpha-I-anichymotrypsin has been found to coexist with this amyloid in Alzheimer plaques. 1 Additionally, a sequence for an inhibitor of the Kunitz family of serine protease inhibitors has been identified which is encoded by a sequence of the beta amyloid protein precursor messenger RNA. 90,126 The protein expressed from an oligonucleotide synthesized from this sequence has been shown to inhibit trypsin in vitro. 67 In fact, this protein may inhibit proteolysis of the precursor. In support of this, cells transfected with the beta amyloid protein precursor that contained the inhibitor sequence produced fewer fragments of the precursor than cells transfected with precursor lacking the inhibitor sequence. 111 The presence of ubiquitin on Alzheimer PHFs also suggests that the proteins comprising neurofibrillary tangles are tagged for cellular proteolysis,18,20 but that the proteolysis was ineffective in degrading these abnormal proteins. At this time, the role of the protease inhibitors associated with beta amyloid protein is still unclear. In fact, completely opposite alternatives still exist. For example, it is not obvious whether inhibiting the proteases that cleave at amino acids 638 of beta amyloid protein precursor would necessarily prevent amyloid formation. Alternatively, lack of protease activity that normally cleaves within the amyloidogenic sequence of amino acids 605 to 638 may be responsible for amyloid accumulation. Obviously, until clarification of these or other alternatives is achieved, therapeutic approaches oriented toward protein inhibitors will remain uncertain. However, precedence does exist for the use of protease inhibitors in numerous other diseases. Thus, once a relevant protease is demonstrated for Alzheimer's disease, it is likely that an inhibitor could be developed relatively rapidly, based on the prior experience of the pharmaceutical industry with this class of drugs. In fact, clinical trials have been (or are being) conducted with inhibitors of the major classes of proteases. For example, captopril, enalapril, and lisinopril, inhibitors of the metalloprotease angiotensin-converting enzyme, have been successful clinically for use in reducing high blood pressure. 88 Four inhibitors of serine proteases (comoslat mesilate, gabexate mesilate, nafamostrat, and urinastatin) have been launched for the treatment of pancreatitis. 4 Alpha 1-PI, an inhibitor of human alpha-I-protease, has recently been launched for the treatment of emphysema. 3 EST. a thiol protease inhibitor, may be useful in blocking proteolysis of myofibrils in muscle atrophy, and is being evaluated in patients with muscle diseases, including Duchenne muscular dystrophy. 57,81 NCO-700, an inhibitor of calcium-dependent neutral protease, is being evaluated for use in blocking proteolysis of myocardial proteins associated with myocardial infarction. 102 In addition, synthetic inhibitors of calcium-dependent neutral proteases may be beneficial to the treatment of osteoarthritis. 10,13,15 Phosphorylation Phosphorylation is a postranslational event that is relevant to the amyloids of both plaques and tangles. Gandy et al. (32) reported that protein kinase C can phosphorylate serine residue 655 of the beta amyloid protein precursor while calcium-calmodulin dependent protein kinase II can phosphorylate this residue and threonine 654 (Fig.2). Since these residues are not within amino acids 597 to 638 of beta amyloid protein precursor which form amyloid, it is not clear how phosphorylation of these sites might affect amyloidogenesis. Gandy et al.32 suggested that that phosphorylation at the protein kinase C site may target the protein for internalization. Saltoh et al.98 reported an elevation of phosphorylation of a 60,000 dalton protein by, postmortem Alzheimer brain samples compared to age-matched controls. The identity of this protein or its effect on cell function is not known. However, the extent of the increased phosphorylation correlated with the number of tangles present but not with the number of plaques or with the presence of gliosis. The PHFs of neurofibrillary tangles do bear phosphorylated sites. These sites have been identified by electron spectroscopic imaging19 and by reactivity with phosphate-dependent antibodies. 23,49,53,56 However, the primary structure of the protein(s) that compnse PHFs remains to be determined. The phosphorylated sites are antigenically shared with several normal cytoskeletal proteins. 8511,106 but these kinases have not been found to phosphorylate the aberrant sites in Alzheimer's disease. The observation that the morphology of the PHF structure is not disrupted by enzyme treatments that remove the phosphorylated epitopes suggests that PHF amyloid is not held together through bonds with these phosphates. 136 Therefore, it is unlikely that simply inhibiting phosphorylation will be useful therapeutically, although it is still possible that dephosphorylation could prove to be a necessary, but insufficient prerequisite for reducing amyloid accumulation. Glycosylation Carbohydrate may be present on the beta amyloid protein precursor, although less evidence exists for its presence on PHFs. Kang et al.61 identified three sequences In the beta amyloid protein precursor that resemble N-linked glycosylation sites, two of which seem likely to be glycosylated in vivo. Fig.2 None of these 3 sites are within amino acids 597 to 638 of beta amyloid protein precursor which form amyloid. The presence of another class of carbohydrate, glycosaminoglycan, on the precursor was suggested recently by Schubert et al.105 who claimed that the size, sequence and antigenicity of the precursor closely resembles a neuronal heparan sulfate proteoglycan. However, these investigators did not eliminate the possibility that proteoglycans were simply inadvertently bound to the precursor in their studies. Moreover, the presence of glycosaminoglycans on the amyloid precursor has yet to be confirmed by others. Nevertheless, synthesis of heparan sulfate proecoglycans has been reported to be elevated two-to four-fold in Alzheimer fibroblasts, suggesting a possible defect in proteoglycan metabolism in Alzheimer's disease. 147 The presence of carbohydrates may affect amyloid formation directly or even indirectly by blocking certain sites for proteolysis as occurs with cartilage-type proteoglycans9,12,29 or by influencing compartmentalization of the precursor to protease-rich sites. The presence of oligosaccharides has been demonstrated on both plaques and tangles by lectin histochemistry71 and by dye-binding techniques. 116,145 However, the actual presence of carbohydrates on PHFs has yet to be demonstrated directly by chemical analyses. If specific carbohydrates are confirmed to be present on Alzheimer amyloids and are shown to play a role in their formation or accumulation, then therapeutic approaches directed toward specific biochemical pathways for carbohydrate metabolism should become apparent. Transglutamination Transglutaminases catalyze reactions that result in the covalent cross-linking of proteins between the glutamine residue of one protein and a primary amino group of another protein (or other compound). 146 Such crosslinks, which increase with age146 would explain the extreme insolubility and lack of protease susceptibility of Alzheimer amyloids110 as well as the increase in the incidence of Alzheimer's disease with age. 45 The cytoskeletal proteins that are associated with PHFs have been shown to be substrates for transglutaminase, although the action of this enzyme does not convert them into morphologically recognizable PHFs. 79,109 To measure the products of transglutaminase reactions, the protein products must first be solubilized and completely hydrolyzed for sequencing. Since their insolubility makes this difficult to accomplish for PHFs, the amount of data regarding this processing event in Alzheimer's disease remains limited. Investigations into these and other posttranslational events that are relevant to aging (including possibly sulfation and carboxymethylation) and Alzheimer's disease [including oxidation of methionine in vascular beta amyloid protein91 remain rich areas for future research. Greater insight about how these events change with age and Alzheimer's disease will likely increase our understanding of any potential role in amyloid formation. 146
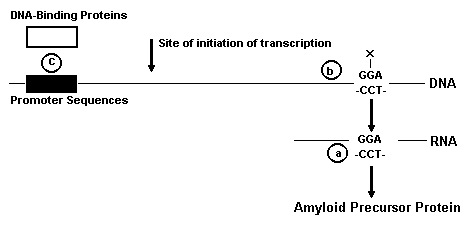
Rationale for Considering Altered Gene Expression in Amyloid Formation Evidence has recently emerged suggesting that an autosomal dominant pattern of inheritance exists for familial Alzheimer's disease (FAD), which comprises a minority of all Alzheimer patients. Through gene linkage studies, St. George-Hyslop et al.117 mapped the FAD gene to chromosome 21, near but distinct from the beta amyloid protein precursor gene. 127,131 Although potentially conflicting results have also been obtained from other groups. 96,104 St. George-Hyslop's observations imply that at least one mutation exists for the FAD gene. If this is eventually confirmed, molecular genetic approaches could then be applied to clone the gene and characterize its function. Clearly, information of this type provides a potential target for gene therapy. However, several assumptions must first be confirmed. The pivotal role of the gene product in mediating the pathology must be established. In addition, whether the normal function of the gene product can tolerate any manipulation of expression without disastrous consequences must also be determined. Another candidate gene for modification of gene expression in Alzheimer's disease is the beta amyloid protein gene. Early reports suggested that an extra copy of this gene was present in Alzheimer patients. 25,26 This finding seemed quite congruous with the situation in Down's syndrome, which results from trisomy of chromosome 21. In Down's patients after about 40 years of age, both of the amyloid lesions of Alzheimer's disease are present. 70 However, several subsequent studies have failed to confirm trisomy of the beta amyloid protein gene in Alzheimer's disease. 31,89,118,124 Nonetheless if accumulation of beta amyloid protein is at least in part due to increased accumulation of precursor, then modifying gene expression to reduce precursor levels might be expected to reduce the formation of beta amyloid protein. On the other hand, it is far too speculative, at present, to propose that the gene for any specific posttranslational processing enzyme would be an appropriate target for gene manipulation. Potential Approaches to Modification of Gene Expression Manipulation of gene expression for therapy is presently a speculative concept. Theoretically, this approach is capable of meeting therapeutic challenges that until now were unapproachable. For this reason it deserves consideration, despite the fact that products of such an endeavor will not become a reality until far into the future. However, the lag time needed to develop this technology into clinically feasible options requires that thought be given now to genes that would be suitable targets. Several approaches to altering gene expression have been investigated. These approaches interfere with different steps of gene expression, as illustrated in Fig. 3. They are designed to manipulate individual genes, as it is not reasonable to assume that nonspecific inhibition of gene expression would provide benefit in any circumstance. Inhibition of gene expression has been demonstrated using antisense RNA oligonucleotides [Fig. 3a; for reviews see (46, 63)]. Gene expression is known to be influenced by regulatory genes. These genes encode for RNA sequences that are complement to the target RNA. The regulatory or antisense RNA binds to its target RNA by base pairing, inhibiting translation of the target RNA. Based on this concept of natural gene regulation, artificial regulation of genes has been achieved using synthetic antisense RNA. For some genes, a 50% reduction in translation has been achieved with antisense RNA.
FIG 3.a A DNA sequence that includes a promoter is illustrated schematically, along with a corresponding RNA sequence and protein translation product. Potential pharmacological intervention sites are numbered. Antisense RNA oligonucleotide sequences act at site a. Antisense DNA oligonucleotide analogs act at site b and generally include chemical modifications (indicated with an X). Altered DNA binding proteins act at site c. See text for further explanation and references. The second approach is similar in concept to the antisense RNA approach. In this case, DNA analogs are synthesized which bind to a segment of the target gene, preventing transcription (Fig. 3b). These analogs are oligonucleotide compounds with modifications to reduce the negative charge of the backbone. 114 Modifications such as these reduce the susceptibility of these compounds to nucleases and enhance their ability to enter cells. Currently, such compounds are being considered for use in AIDS. 44,76,80 The third approach involves manipulation of DNA-binding proteins that regulate transcription (Fig. 3c). These proteins are termed promoter-specific transcription factors [for reviews, see (27, 121)]. They interact with DNA at distinct DNA sequences (termed promoter regions) that are upstream from the site of initiation of transcription. Interactions of these proteins with DNA promoter regions regulate transcription. Such interactions selectively allow the components of the transcription apparatus, such as RNA polymerase II, to initiate transcription. The selectivity results from the specificity of DNA-binding proteins for specific DNA sequence elements in the promoter region. Some sequence elements theoretically may be specific for particular genes. Recently, the promoter region for the beta amyloid protein gene has been described, as well as several potential promoter specific transcription factors. The promoter region that has been identified has features that are typical for promoters of housekeeping genes. 99 Possible transcription factors include AP-1,99 acute phase proteins,73 nerve growth factor,73,82 and interleukin-1. 43 The amino acid sequences of DNA binding proteins have been altered in an effort to change the DNA sequence that these proteins recognize. Such changes have indeed resulted in altered gene expression. 133,134 Green and Chambon47 reported altering gene expression by changing estrogen and glucocorticoid receptors, which serve as transcription factors. They produced a hybrid protein of the 2 receptors that activated expression of a glucocorticoid-inducible gene in the presence of estradiol. The foregoing discussion provides an example of how DNA-binding proteins can be altered to modify their ability to regulate expression of their target genes. As the genetic mechanisms involved with amyloid accumulation become more clearly elucidated, the DNA binding proteins that specifically regulate the expression of these genes may be useful as potential drug targets.
The recent increase in information regarding the molecular pathology of Alzheimer's disease suggests it should be feasible to develop therapeutic approaches directed toward the abnormal protein accumulation present in the brains of these patients. In fact, some precedence for developing clinically effective compounds that alter amyloid deposition already exists for several amyloidoses associated with other diseases. In Alzheimer's disease, therapies directed toward posttranslational processing and gene expression currently provide two of the more promising approaches. In addition to possibly providing a successful treatment, agents emerging from these general approaches may also provide invaluable tools to probe the role of amyloid accumulation in Alzheimer pathology, thus increasing our fundamental understanding of the disease. The key impediment to employing these approaches today is the lack of sufficient knowledge regarding the specific molecular mechanisms responsible for amyloid formation in Alzheimer's disease. As progress in this area continues to be achieved, a more focused approach to therapeutic intervention should emerge, eventually leading to ways of effectively reducing the classic neuropathology associated with Alzheimer's disease. The impact that removing these pathological lesions may have on the neurodegenerating consequences of Alzheimer's disease, or on the clinical status of its victims, will then likely become the intense focus of the next generation of research in this area. Source: This article was printed in "Microbiology of Aging, Vol. 10, pp. 451-461. ©Pergamon Press pic. 1989. Permission was granted by the publisher to transcribe this article for the Internet by DMSO Organization. Copyright is retained by the publisher. No part of this article may be reprinted without written authorization from the publisher.
|
|||
© 2001-2022
All rights reserved |