|
||
DMSO Background Literature |
|
|
The Amyloid Proteins of Alzheimer's Disease as Potential
Targets for Drug Therapy, Part 1
Claudio B. Caputa and Andre I. Salama Department of Pharmacology • ICI Pharmaceuticals Group • ICI Americas Inc. • Wilmington, DE 19897 Received 21 February 1989 Abstract CAPUTO. C. B. and A. I. SALAMA. The amyloid proteins of Alzheimer's disease as potential targets for drug therapy. NEUROBIOLOGICAL AGING 10(5) 451-461, 1989.--Two amyloid proteins accumulate in Alzheimer's disease. These proteins, beta amyloid protein and paired helical filament protein, are present in the hallmark lesions of Alzheimer's disease, neuritic plaques and neurofibrillary tangles. Although the amino acid sequences of these two proteins are likely to be different, they nevertheless share certain physical charcteristics which define each as belonging to a common class of proteins, amyloid proteins. Since these proteins are probably important in the pathology of Alzheimer's disease, drugs that prevent their accumulation should have therapeutic utility. Based on the amyloidosis associated with other diseases, three mechanisms for amyloid formation have emerged. These mechanisms form a framework for studying Alzheimer amyloids and designing intervention. One mechanism involves posttranslational events which render a normal protein amyloidgenic. Proteolysis, phosphorylation, glycosylation, and transglutamination may be relevant posttranslational events in Alzheimer's disease. If more conclusive evidence can be generated suggesting that these events are involved in the abnormal formation of amyloid in Alzheimer's disease, then these events will become viable targets for drug therapy. Another mechanism for amyloid formation results from expression of an abnormal gene which, in the case of familial Alzheimer's disease, may be an important etiological component. A third mechanism involves the accumulation of a normal protein to a threshold concentration that spontaneously forms amyloid. An effective therapeutic approach for these last two mechanisms could include pharmacological manipulation of gene expression. There is no means presently available to treat the pathogenesis of Alzheimer's disease. However, insight gained from recent research efforts is beginning to provide the empirical foundation required to develop logical therapeutic approaches. In fact, because the increased accumulation of amyloid proteins is now recognized as an important pathogenic event of Alzheimer's disease, the successes and failures gained in attempting to treat other forms of amyloidosis may provide some clues for treating the particular CNS amyloidosis associated with Alzheimer's disease. Amyloid is a term that refers to a class of proteins that share several properties. These proteins have a beta pleated sheet structure, which has a high affinity to bind Congo red, produces birefringence in polarized light, and produces a characteristic X-ray diffraction pattern. 35,37,64 They are insoluble and are not susceptible to proteases. 110 A number of proteins with unrelated amino acid sequences share these properties. 36 It is likely that at least two different proteins which share the properties of the amyloid class accumulate in Alzheimer's disease. 138 One accumulates extracellularly, both in brain parenchyma (where it is associated with neuritic plaques) and in the walls of blood vessels. This protein is called beta amyloid protein and its primary structure has been defined. 140 A second protein with the general properties of the amyloid class accumulates intracellularly in neurons of Alzheimer brains. 16,36 The accumulation of this protein is visible by light microscopy as neurofibrillary tangles. The tangles are composed of structures that appear to be paired helical filaments (PHFs) by electron microscopy and differ in morphology from the fibrils formed from the beta amyloid protein found in plaques. 130 PHFs are present in tangles as well as within some of the neurites that form neuritic plaques. PHF-containing neurites are found in the periphery of the plaque, whereas deposits of beta amyloid protein form the central core of mature plaques. In contrast to the beta amyloid protein. the primary structure and the number of proteins comprising PHFs are unknown. Various proteins have been proposed as components of PHFs. The microtubule-associated protein tau has been demonstrated in PHFs by immunohistology48, 84, 141 and by molecular biological studies. 4O, l37 Neurofilaments, the microtubule-associated protein MAP2, vimentin, as well as the beta amyloid protein of Alzheimer plaques have also been suggested to be present in the PHF structure. 68, 74, 83, 119, 143, 144, 148 The possibility that the two primary lesions of Alzheimer's disease (plaques and tangles) may share an identical abnormal protein, beta amyloid protein, has attracted particular attention by investigators interested in the pathogenesis of the disease. However, the presence of beta amyloid protein in tangles still remains controversial. 16 For the purposes of this review, PHFs will be considered as an amyloid protein distinct from the beta amyloid protein of plaques. Identification of the cell(s) that produce Alzheimer amyloid proteins is addressed in other reports in this issue [Ivy Street Note: these reports are not available at this site]and thus will not be addressed in this review.
The Role of Amyloid Proteins in Alzheimer's Disease Amyloid proteins with amino acid sequences that are distinct from those of Alzheimer's disease accumulate in numerous diseases. The presence of these insoluble proteins almost invariably leads to the loss of function and death of the cells in which they form. 35, 36 It seems generally true that the presence of amyloid is incompatible with normal cell function and survival. Although definitive proof may still be lacking and disagreement among investigators still exists, considerable circumstantial evidence has been generated to support an important role of amyloid in the ceullar neural pathology of Alzheimer's disease. To begin, the presence of large numbers of amyloid-containing plaques and tangles is used as a definitive diagnosis of Alzheimer's disease. 6 They not only exist in all confirmed cases of Alzheimer's disease, but there is evidence to support the concept that the numbers (above threshold levels) of senile plaques and neurofibrillary tangles present in an individual correlates well with the extent of dementia. 135 Plaques and tangles accumulate in highest numbers in the hippocampus and the neocortex. 97 Functionally, these regions play important roles in memory and cognitive function. These functions are particularly compromised in Alzheimer patients and represent the hallmark symptoms in the earlier stages of the disease. Memory processes in the hippocampus and cortex are mediated through the neurotransmitter, acetylcholine. 5 This neurosmitter becomes depleted as Alzheimer's disease progresses. 7, 24, 86 In fact, the extent of the deficit in cholinergic neurochemical parameters also correlates well with the numbers of plaques and tangles present. 87 It is conceivable that, as insoluble proteins, the presence of extracellular amyloid proteins in Alzheimer brains would disrupt neuronal function. The extracellular deposits of amyloid create physical barriers between neurites. The neurites involved in plaque formation show numerous degenerative changes, which may be compromised further by amyloid deposits that impede normal synaptic structure and probably function. In addition, recent preliminary evidence suggests that amyloid peptides may have specific toxic effects on neurons. When cDNA corresponding to the carboxyl terminal 105 amino acids of the beta amyloid protein precursor was transfected into neuronally differentiated cells, degenerative changes were observed. 142 In contrast to the effects of these fragments, the deposited fibrils of beta amyloid protein do not seem to be neurotoxic, as amyloid deposits are found in the absence of dystrophic neurites. 139 In fact, there exists a negative correlation between the number of dystrophic neurons and the density of amyloid fibrial deposits. 138 lntracellular amyloid accumulation has also been implicated in neuronal dysfunction in Alzheimer's disease. Electron microscopy of neurons that contain intracellular PHFs has led to the suggestion by Saper et al.101 that such an accumulation of PHFs may disrupt the cell and cause its death. The PHFs that remain after all cellular organelles are lost provide a tombstone of these cells. In support of this idea, Sumpter et al.122 demonstrated that cells with extensive neurofibrillary tangle involvement have little useful cytoplasm and few ribosomes compared to less involved and noninvolved cells. From this discussion emerges the likelihood that insoluble amyloid protein, which compromises PHFs accumulates within Alzheimer hippocampal and cortical neurons, creating functional disturbances and eventually cell death. Moreover, the increased extracellular accumulation of beta amyloid protein in selected brain regions also likely hinders the function of these particular brain regions. Thus, this brain amyloidosis is likely a contributing factor to the cognitive pathology observed in Alzheimer's disease. 72, 138 The specificity of plaques or tangles to Alzheimer's disease is important when considering whether these lesions are a common consequence of neurodegeneration. These lesions do occur in other diseases. However, plaques that are made up of the same beta amyloid protein as in Alzheimer's disease have been confirmed only in Down's syndrome and hereditary cerebral hemorrhage with amyloidosis of Dutch origin (HCHWA-D). 21, 75, 92, 132 Plaques in HCHWA-D brains are not accompanied by tangles. Tangles also occur in a number of diseases and in several experimental conditions in animals. However, the filaments that make up these tangles in most cases are not morphologically similar to PHFs. 100 Two notable exceptions are the Parkinson's dementia of Guam and Down's syndrome. 107 Tau protein antisera recognize these tangles as well as tangles from Alzheimer's and other neurodegenerative diseases. 59 In certain experimental axonopathies it is known that the protein that forms the insoluble filaments of tangles is neurofilament. 103 However, these filaments are morphologically distinct from the PHFs of Alzheimer's disease. Alzheimer tangles differ in other ways, as well. Sulfated glycosaminoglycans are associated with Alzheimer tangles but not with tangles of progressive supranuclear palsy, Pick's disease, subacute sclerosing panencephalitis, and postencephalitic parkinsonism. 116 Although PHFs are not specific to Alzheimer's disease, it is premature to conclude that the formation of neurofibrillary tangles composed of PHFs is a common consequence of neurodegeneration. From the foregoing discussion emerges the concept that amyloid accumulation in Alzheimer's disease causes a loss in function of the affected cells. Given this, it follows that preventing the progress of amyloid deposition very early in the disease process may be therapeutically useful, at least until another pathological process--potentially initiated by the same ecological event--takes over. Additionally, one could hypothesize that by preventing amyloid deposition, the presence and nature of these underlying pathological events can be identified. An advantage of this approach is that it can be undertaken without any knowledge of the root etiology of Alzheimer's disease.
Current Obstacles to Controlling Amyloid Accumulation Several impediments have thus far hindered progress in pharmacologically treating Alzheimer amyloid deposition. For example, the mechanism of amyloid formation in Alzheimer's disease is unknown, creating severe difficulties in designing agents that intervene in this process. In addition, no animal models for brain amyloidosis with beta amyloid protein deposits or PHFs exist, creating difficulties in testing any potential intervention. Although there exist several ways to generate amyloidosis in animals (i.e.. scrapie infection), the amyloid protein produced is not the same as the beta amyloid protein found in Alzheimer's disease66 and no PHFs form. Another obstacle to controlling amyloid accumulation is that neither the nature of the precursor of the PHF protein nor the essential processing events that convert soluble proteins into Alzheimer amyloids are known. Current research is intensely addressing these issues. In the meantime, addressing any one particular processing event runs the inherent risk that it is an epiphenomenon which occurs after or independent of amyloid deposition. In fact, the possibility remains that the whole process of amyloidosis itself may be an epiphenomenon of Alzheimer's disease, in which case effective intervention of amyloid deposition may have no effect on the consequence or progression of the disease. For example, the cytoskeletal changes in Alzheimer's disease suggest that an extensive disconnection syndrome is present that effectively isolates the hippocampus from the limbic and association cortices. 108 Of course, any effective intervention of amyloid deposition would test this hypothesis of amyloid as an epiphenomenon. A related obstacle is that amyloid deposition may not represent the initiating event in plaque formation. For example, a common type of plaque in monkeys contains enlarged neurites but no beta amyloid protein deposition. 120 "Primitive" plaques in Alzheimer's disease also lack a central amyloid core. 139 Furthermore, diffuse plaques are present in Alzheimer brains which contain amorphous deposits of immunoreactive beta amyloid protein that do not stain Congo red and are no( associated with dystrophic neurites. 111 These diffuse plaques may develop into more classical neuritic plaques with amyloid cores with time or within certain regions of the brain. PHFs appear within these enlarged neurites of plaques,139 but whether they precede the appearance of beta amyloid protein deposits in those plaques that contain these deposits is not clear. What is clear, however, is that there must be additional underlying events that lead to amyloid deposition as well as many other pathological changes. The success of therapy aimed at amyloid deposition will be determined in part by the specific pathology these other changes produce. The question of which Alzheimer amyloid protein, either PHFs or beta amyloid protein, should be the target for therapeutic intervention is difficult to answer at present. In general. the current lack of understanding of the biochemical relationship between plaque formation and tangle formation prevents one from choosing either as a more appropriate target. Even though there is no confirmed biochemical relationship between the two Alzheimer amyloids, one group reported that the amino acid sequence for PHFs matches beta amyloid protein. 74 If these two amyloids are comprised of the same protein, then preventing deposition of beta amyloid protein might be expected to reduce the formation of both plaques and tangles. On the other hand, assuming PHFs are comprised of an amyloid protein other than beta amyloid protein, it is not clear at present whether it is necessary to have amyloids accumulate to produce dementia. Thus, it remains possible that reducing only one of these amyloids will merely delay the dementia produced by the other amyloid. This possibility seems reasonable since dementia occurs in patients with PHFs in tangles but with no plaques present, such as in Guamanian Parkinsonian dementia,50,55 as well as in patients with plaques but no neocortical tangles. 129 Until the relationship between plaques and tangles is understood, the possibility remains that blocking the formation of PHFs in enlarged neurites may not impact plaque development. In fact, a crucial question that remains is whether one can improve the function of brain regions with enlarged neurites simply by reducing beta amyloid protein formation. Although the complications discussed above make an amyloid approach to Alzheimer's disease therapy more difficult, a number similar obstacles have been overcome in treating other amyloidosis, thus providing a framework to structure analogous approaches for Alzheimer's disease.
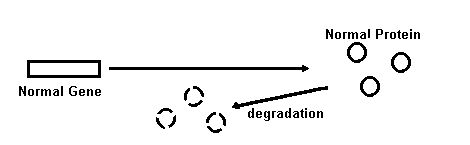
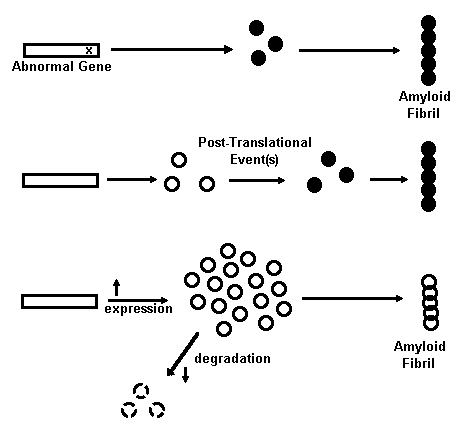
Possible Insight for Treatment of Alzheimer Amyloids Based on Common Characteristics of Amyloidoses Control Situation: No Amyloid Formation Mechanisms of Amyloid Formation Fig. 1. Mechanisms for amyloid fibril formation are shown schematically. Open circles represent normal protein molecules, solid circles are amyloidogenic protein molecules, and overlapping circles are the fibrils made from these proteins. See text for an explanation of each mechanism and references. Amyloidosis is a pathological event that is manifested in a number of diseases and has been studied extensively [for reviews, see (16, 35, 36, 66)]. From these studies a number of common features of all amyloidosis have emerged, despite the fact that amyloids in different diseases are deposited in different tissues for different reasons and are formed from various unrelated proteins. 16,35,36,66 An understanding of the features they share in common, however, is likely to provide insight into the process of amyloidosis in Alzheimer brains and its eventual treatment. The molecular mechanisms that convert a soluble protein into insoluble amyloid protein are, for the most part, unknown. It has been proposed by Wisniewski139 that a two-step process is involved. The first step involves the production of a normal precursor protein and the second involves modification of the protein to a form that yields amyloid. Four mechanisms for shifting this kinetic process toward the direction favoring amyloid production have been proposed,16 as illustrated in Fig. I. The first mechanism is a mutation that results in an altered primary structure of a protein which is more amyloidogenic than the nonmutated version. Examples of diseases that result from this mechanism include familial amyloidotic polyneuropathies and cerebral hemorrhage with amyloidosis of Icelandic type. 16,33,34 Although no data has emerged to support this possibility for the amyloid gene per se, it continues to attract attention as an important mechanism for that segment of Alzheimer's disease bearing an hereditary etiology, termed familial Alzheimer's disease (FAD). 127,13 The second mechanism proposed for amyloid formation is the occurrence of a posttranslational modification of the precursor protein that renders it amyloidogenic. This mechanism has been implicated in some types of amyloidoses. Examples include nodular pulmonary amyloidosis, amyloidosis associated with multiple myeloma, light chain disease, and some secondary amyloidoses with amyloid fibers composed of a protein termed A protein (a peptide that is antigenically related to an acute-phase serum component). 35 As discussed later, this mechanism remains a strong possibility for the accumulation of the beta amyloid protein in plaques, and possibly the amyloid of PHFs as well. The third and fourth mechanisms both result in the accumulation of a precursor protein to a threshold concentration that favors amyloid product formation. 16 An increased rate of precursor synthesis would cause this effect as occurs in light chain and secondary amyloidoses. A decreased rate of precursor degradation would also cause this effect. Familial and acquired amyloidoses have been suggested to result from this mechanism. 16 Although little or no direct evidence exists for these mechanisms in Alzheimer's disease amyloidoses, each nevertheless remains a viable possibility for both types of Alzheimer amyloid formation. Besides the possibility that amyloid formation in different diseases may share common mechanisms, the amyloid deposits from different diseases also share certain biochemical properties. The glycoprotein, amyloid P component,22 glycosaminoglycans,115 and elastase113 have each been reported to be associated with all amyloids examined, including those in different tissues and those comprised of different amyloid protein precursors. Calcium-dependent interactions between serum amyloid P component (a precursor of amyloid P component) and glycosaminoglycans have been demonstrated. 51,52 The roles of these factors in amyloid formation are unknown. Should any of them be shown to be essential in amyloid formation, they may provide another site to target intervention. Despite a lack of knowledge of the mechanisms involved in various clinically significant amyloidoses, several reports of effective treatments have emerged, Dimethyl sulfoxide, and amyloid fibril denaturing agent, blocks amyloid formation from Bence Jones proteins in vitro. 58 Dimethyl sulfoxide and colchicine each reduce amyloid deposits in casein-induced amyloidosis in mice and have generated intriguing results in human amyloidosis, as well. 36,58,62 For example, colchicine treatment has been reported to reduce the size of renal amyioid deposits and induce clinical remissions in several cases of familial Mediterranean fever and amyloid nephropathy. 94 Additionally, prophylactic administration of colchicine has been suggested to prevent amyloidosis in patients with this disease. 41 Dimethyl sulfoxide may also have clinical utility in patients with amyloid A systemic amyloidosis and lichen amyloidosis. 8 Alpha-chymo trypsin and bromelain, proteases that are used as oral mucolytics, and collagenase may be partially effective in cases of familial amyloidotic polyneuropathy. 2 Finally, oral etretinate therapy has been reported to be effective in some cases of lichen amyloidosis, with no remaining amyloid present in those biopsies that were examined. 54 Etretinate is a retinoid that is effective in a number of keratinizing conditions. In contrast to Alzheimer amyloid, other amyloids can be induced to form in animals. Casein administration to mice induces amyloidosis; a mutation in the protein apo A-II induces senescent amyloidosis in mice; scrapie, which is a disease that is tranmissible to mice, hamsters and sheep, is accompanied by amyloid plaques in the brain; and light chain amyloid has been induced in cell cultures. 66 Such animal models have provided opportunities to assess methods of chemical intervention for certain types of amyloidosis. It has yet to be established whether the underlying mechanisms for the actions of dimethyl sulfoxide and colchicine will bear any relevance to Alzheimer amyloidosis. The fact that both animal models as well as certain therapeutic approaches exist for certain forms of amyloidosis offers encouragement that it may also be possible to develop logical treatment approaches for the pathogenesis of Alzheimer's disease.
Source
This article was printed in Microbiology of Aging, Vol. 10, pp. 451-461. © Pergamon Press pic. 1989. Permission was granted by the publisher to transcribe this article for the Internet by DMSO Organization. Copyright is retained by the publisher. No part of this article may be reprinted without written authorization from the publisher. |
|
© 2001-2022
All rights reserved |